Considerations for Creating Plain Language Summaries
Article
薬事規制関連支援サービス

By Darcy Grabenstein
According to a go-live plan released in October by the European Medicines Agency, its new Clinical Trial Information System (CTIS) is on track to launch Jan. 31, 2022. Along with the new registry portal comes application of European Clinical Trial Regulation , which specifies what documents must be disclosed on the portal and when. The European Medical Writers Association (EMWA) cites that plain language summaries (PLS) of clinical trial results, “following health literacy and numeracy principles, will also need to be posted on the portal, detailing in lay language how the trial was conducted and its results.” Additionally, the UK’s Health Research Authority (HRA) requires a PLS within 12 months of study completion for trials with sites in the UK that end on or after Sept. 15, 2021.
Given that writing PLS can be a lengthy undertaking — especially if translations or a patient review panel are called for — sponsors would be well advised to start the process now. “Sponsors should not be caught unaware of what goes into this process,” says Courtneay Parsons, Head of Plain Language Summaries at ClaritiDox. “You’re not going to just sit down at a laptop, start writing, and be done a few days later.”
While many medical writers are well versed in writing the Informed Consent Form (ICF), which in theory should be written in plain language, in reality the ICF is more of a lengthy legal document that does not typically meet PLS standards. Contracting a dedicated PLS writer can save time — and headaches. A PLS writer is skilled in making study results understandable, but without sacrificing accuracy. The person who drafts your PLS needs a fluent knowledge of PLS requirements, of source documents, and of what efficacy and safety data need to look like in both. Without it, Parsons says the PLS process will be “like walking through a mine field.”
Thomas Wicks, Head of Transparency for Citeline, stresses that advance planning is key to streamlining PLS. “You should incorporate plain language concepts throughout all your documents that eventually will be disclosed. For example, if you include plain language elements into your protocol, they can be used for consistency in other documents.”
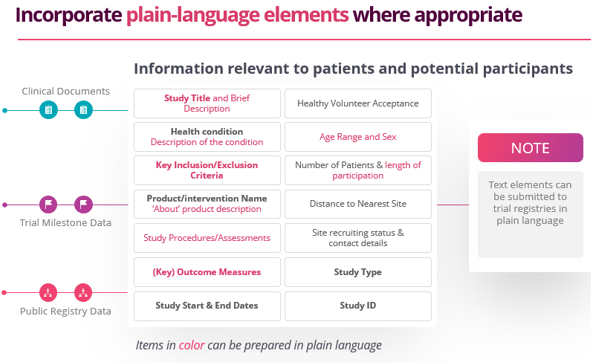
Source documents for PLS include:
Parsons warns that sponsors must not choose to include information dependent on which results look good. This is known as “cherry picking” and is unacceptable because it could be considered promotional and puts the product ahead of patient information needs. Sponsors must be objective in their presentation of data, using standardized definitions and methodology on what efficacy data to include and why.
Creation of a PLS template is recommended, as stated in the Good Lay Summary Practice (GLSP) guide adopted by the Clinical Trials Expert Group (CTEG), a working group of the European Commission representing Ethics Committees and National Competent Authorities (NCA). A strong template can be applied to all plain language summaries of trial results, with built-in guidance to help teams adapt to specific situations, as needed.
A template should take into consideration the following information:
One of the most common issues faced by sponsors in creating PLS is identifying what Tables, Listings, and Figures (TLF) they will need that are not already included in a standard TLF suite. When patient case report forms are collected and finalized, the data are put into by-patient listings and summary tables. Historically, a TLF suite for a trial contains everything needed to write the clinical study report (CSR) for that trial. However, clinical trial registries, and now PLS, require data outputs that are not necessarily included in every CSR’s TLF suite.
For example, the EU CTR and associated GLSP guidance discuss safety endpoints in the context of adverse reactions, defined for the PLS as “adverse events for which the investigator has indicated at least a reasonable possibility of an established causal relationship between the event and the [investigational medicinal product] based on an analysis of available evidence.”
As a result, there are often safety variables needed for a PLS ― such as the percentages of patients by treatment arm who discontinued treatment due to related adverse events ― that are not often included in a sponsor’s TLF package. What’s more, the GLSP recommends presenting serious related events and nonserious related events separately (i.e., stripping serious related events from the counts of any related adverse events): “The EU Expert Group on Clinical Trials Recommendations suggest that the serious adverse reactions be listed first in the LS, followed by the ‘other,’ common adverse reactions listed by frequency. Clear separation of serious adverse reactions from the latter category is intended to avoid duplication of information within the PLS.” Virtually no sponsors have TLFs set up to do this, says Parsons.
Before we delve into what reviewers should be looking for, let’s outline who should be on the review team. Ideally, reviewers will include some of the following stakeholders representing a cross-section of the organization, those with specific expertise to provide an internal system of checks and balances:
To enable reviewers to work most effectively, they must be given context as to what they are reviewing. A PLS will look unlike any document they have seen before, including the ICF, which must be in plain language. A PLS goes the extra step, because the intended audience is the general public, not merely those who have the specific disease or condition.
Reviewers should be given a heads up that graphics and colors will be used that they are not accustomed to. Legal/regulatory reviewers should be advised that PLS are written at a 6th- to 8th-grade reading level, a far cry from typical clinical documentation. Setting expectations up front minimizes reviewers’ efforts and enhances overall collaboration with the team. Sponsors should formalize reviewer training materials to streamline the process across teams.
A PDF of the PLS can be uploaded to CTIS, but this format is not optimal for the visually impaired. Of course, PLS can be sent directly to patients either digitally (consider an audio file) or via traditional mail, but sponsors may encounter logistical and budgetary constraints. For pediatric trials, sponsors may want to consider providing a video version of the PLS.
Sponsors uploading PLS to their own clinical trial websites should take into account online accessibility issues. Larger font sizes and alt text for images are two simple ways to accommodate visitors of varying abilities. In addition, website plug-ins are available that can transform content into black and white, set a high contrast, and automate other adjustments for increased accessibility.
In the ClinicalTrials.gov Protocol Registration and Results System (PRS), the fields for brief title and summary have notes that specifically state they are mean to be authored in “patient-friendly” text, though only a handful of sponsors are doing so, notes Parsons.
The key to increasing access to PLS is making it available in a variety of formats, in a variety of locations, in a language that educates, not intimidates.




