Protecting Data in Clinical Trial Disclosure: Managing Commercially Confidential Information (CCI)
Article
Regulatory & Compliance

In January 2023, the European Medicines Agency (EMA) began requiring all new clinical trials to be registered through its Clinical Trials Information System (CTIS), replacing the EU Clinical Trials Registry.1 CTIS is now the online system for regulatory submission, authorization, and supervision of clinical trials in the European Union and the European Economic Area.2
There are two restricted and structured workspaces within CTIS. Sponsors and marketing authorization applications can access the sponsor workspace. The authority workspace is accessible to the national competent authorities of the EU member states, ethics committees, the European Commission, and the European Medicines Agency (EMA).2
CTIS also has a public website that can be accessed by any user without the need to register. These users will be able to search the website and find information on all clinical trials.
Because of the existence of this public-facing site, CTIS allows sponsors to submit two versions of various documents. One version is meant for publication on the public site, while the other is not for publication.3
In the public-facing version of documents, all information submitted to CTIS will be made available to the website unless it’s information that needs to be protected. According to the EMA2 this includes:
In September 2023, the EMA relaunched the Policy on the Publication of Clinical Data for Medicinal Products for Human Use, or Policy 0070 for short, after suspending it during Brexit and the COVID-19 pandemic. The goal of the policy is to increase transparency and promote the sharing of clinical trial data to benefit health for all.
As part of Policy 0070, marketing authorization holders (MAHs) are required to anonymize all personal data that could identify patients participating in the trial. The policy also allows for the redaction of certain data that are deemed CCI, or “any information … that is not in the public domain or publicly available and where disclosure may undermine the legitimate economic interest of the applicant/MAH.”
It can be unclear during the redaction and anonymization process to determine what data is CCI. While the EMA offers guidelines as to what types of information may qualify as CCI, sponsors typically expect to redact a broader scope of data as confidential. The EMA it does not consider the following to be CCI:
In the recent Citeline webinar, “How to Protect Sensitive Data in Public Clinical Trial Disclosures: Industry Insights and Best Practices for Data Anonymization and CCI Redaction,” panelists discussed best practices for redacting CCI in clinical trials.
Greg Shear, disclosure services lead at ClaritiDox, has worked in clinical trial disclosure since 2015 with prior experience in clinical research and technical writing. He specializes in trial registration, results, reporting, document anonymization, plain language summaries, and data sharing.
Shear says the short answer to what data the EMA will consider CCI includes manufacturing details, quantitative composition, future development plans, innovative analytical methods or new biomarkers, and site facility and equipment information.
“More broadly than that, it also depends on if you’re deferring the publication of your documents or not, because we’ve seen that member states have been more broad in terms of allowing certain information to be redacted if you publish those documents sooner,” he says. “But if you’re going for the combination of deferring the document publication to later and including redactions that are out of scope of these eight fields that are given or any type of extensive redaction, you’re likely to get pushback.”
Shear says he has worked with sponsors that want to redact information beyond what is cited in the guidance. “They might want to redact PK [pharmacokinetic] information or the structural information or metabolic pathways or preclinical trial doses or anything like that,” he says. “But again, if you’re trying to do extensive redaction like that along with the deferral, you risk getting pushback in the form of an RFI [request for information], which could lead to having to unredact a lot of that information later on.
“I think it is a bit of a risk/benefit analysis in terms of determining how valuable that information is to your company to keep it protected and are you able to adapt to either a very short turnaround time and unredacting or revealing that information or possibly having that being put out even if you determined that was something that was sensitive.”
Elliot Zimmerman, CEO, Real Life Sciences, is passionate about accelerating trials and making it easier to share results. He serves on the steering committee of the Clinical Research Data Sharing Alliance (CRDSA), a multi-stakeholder consortium with the goal of accelerating the discovery and delivery of therapies to patients by expanding the research value of data collected through the clinical development process.
Zimmerman says with the relaunch of Policy 0070, regulators are expecting CCI to be thought through much sooner in the cycle.
“Now in your pre-submission meeting, you’ll need to bring to the table your approach and outline for what you’re categorizing as confidential information so that can be reviewed by the regulator very early in the process,” he says. “And the requirement for justification tables for each of the respective CSRs [clinical study reports] and documents that are a part of Policy 0070 will still need to be defined. Those tables will need to be created and submitted with your proposal package and your final package as well.”
Shear cautions there may be different definitions of what’s acceptable as CCI between Policy 0070 and CTIS. “For CTIS, you’re talking about all of the different member states in the EU,” he says. “Just because something that was out of scope was accepted on one submission doesn’t guarantee it’s going to always be accepted.”
As far as best practices for identifying, minimizing, and redacting CCI, Shear says applicants should start by getting relevant stakeholders like clinical study leads, regulatory affairs, intellectual property and patent, and manufacturing involved in looking at documents while they’re still being drafted. They should then identify product terms that aren’t publicly disclosed anywhere and, if they get out, will have a reasonable commercial impact to either hurt the applicant or help a competitor.
“I think that’s the best strategy because if you can identify where the terms are and you could pre-plan that justification, you at the very least have a response to the national authorities if they give you an RFI,” he says. “And if that information is still confidential, by the time you get to a marketing submission you already have that justification pre-written out and you could copy/paste it into the template.
“I think the biggest thing is don’t expect anyone outside your organization to be able to tell you what your CCI is because someone like me, I could read through the regulations and tell you what might be allowable. But there could be many things specific to your product that only are known through your own development or your future strategy that someone is going to have to find out and at least put in a list somewhere so that you can look for those redactions.”
Shear says once the list of terms is prepared, sponsors should tell the medical writing teams to minimize or use more vague language in the documents whenever possible. Also, they should try to keep CCI out of anything that needs to be entered into a main characteristic field like objectives. “If you list something in your objectives that has to be pasted into a CTIS field, you can’t redact that,” he says.
“Ultimately, I just want to urge everyone to just be selective because if you’re reducing the number of redactions and you’re truly looking at what fits the bill as CCI, you’re reducing your risk of an RFI, and it’s less work for you later on. If you have a substantial modification and you have a new version of that document and if you are deferring these documents, you should be extra careful to make sure you’re only redacting things that are going to be CCI at the end of that deferral period because they urge you to be time dependent.
“Lastly, just be consistent across all your trials, which is another reason to have this one library for every product to know that ‘every trial we submit, we’re not going to include that term. We’re not going to include that one piece of the manufacturing process or anything like that.’”
“One tactic that we’ve seen be really helpful is prioritizing the most dense clinical documents first from a redaction perspective,” Zimmerman says. “For example, start with the IB [investigator’s brochure] because if you can solve for the IB in terms of making your CCI and redaction decisions, then all those decisions made in the IB will flow down to the other documents.”
Zimmerman notes that cross-functional team training is also important because there may be people who have never been involved in a CTIS project. “Having a very structured kickoff meeting with an education component and real-life examples of what is and is not CCI and why will go a long way in helping to ramp up that team so that it’s a much more productive discussion when you get into the decision-making process for your project,” he says.
Zimmerman says there are numerous risks to poorly managing data anonymization and redaction.
“One would be if you’re talking, for example, about a Health Canada or EMA Policy 0070 submission, the regulators do review this very closely and they will critique it and they will kick it back for adjustments,” he says. “So, it’s really critical you have alignment with the regulator early in the process through your pre-submission meeting or the PIM meeting with Health Canada.
“That said, if it’s not done well, at a minimum you’re going to have delays in your submission. And delays mean time and money and resources, which could result in non-conformance notifications from the regulator. And then there’s a public perception around, ‘Well, this is required and it’s not happening in a timely manner. Why isn’t this particular organization able to publish this information as deemed necessary from a regulatory perspective?’
“Being consistent in your approach and your strategy for anonymization for these submissions is important as well, so that the reader, whether that’s an academic researcher or a lay person, knows what to expect from your organization and they’re going to find that in a consistent manner. So that could be a risk as well if it’s not done consistently, that they would struggle to be able to interpret it.”
”According to Zimmerman, there’s also a risk of delaying the study if the redactions aren’t being managed in a streamlined manner. “And there are huge implications of that financially and operationally,” he says. “Having a really well-oiled machine in terms of the handoffs between teams, the approval process internally around CCI, all of these things … really do have a direct impact on allowing that study to progress through the CTA [clinical trial agreement] process. And getting through that CTA so that the next phases of the trial can actually continue is maybe obvious but worth stating.”
Figure 1: Main Challenges in Redacting CCI
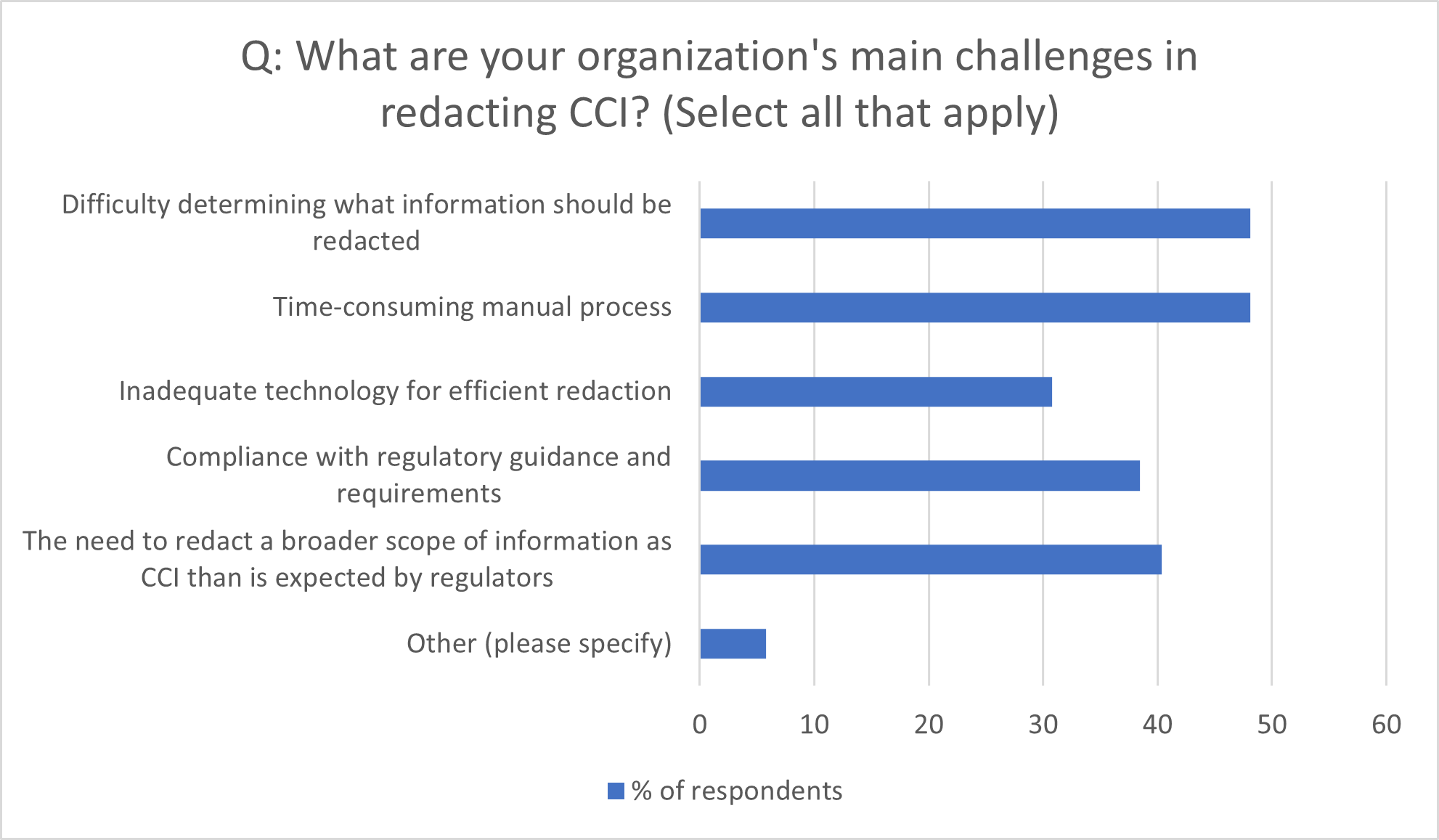
Source: Citeline survey data, July-October 2023
Figure 2: Primary methods for redacting CCI from CTIS documents
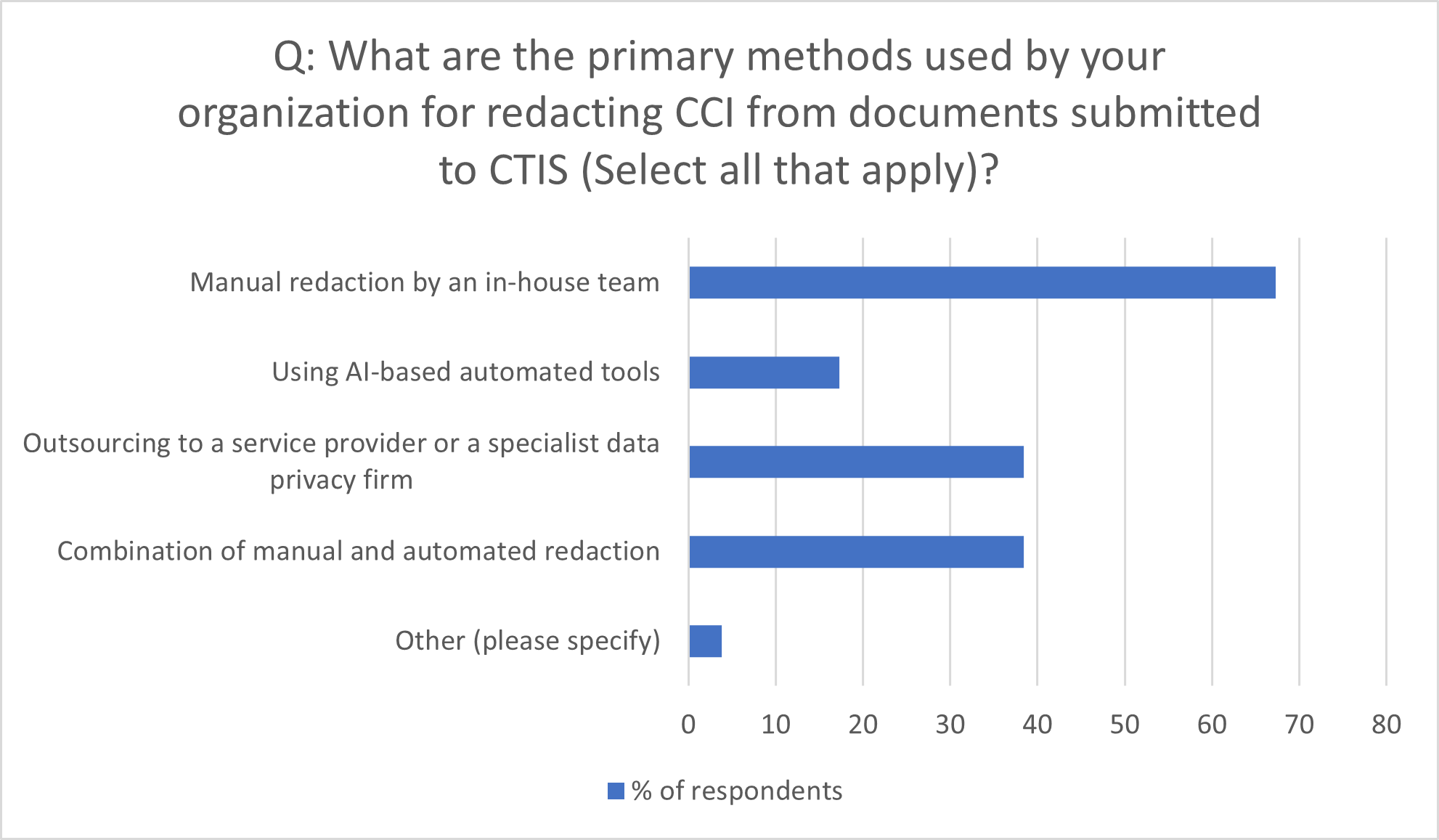
Source: Citeline survey data, July-October 2023
Shear says it can be a challenge to conduct redactions across global regions.
“Definitely from at first gathering, where you are submitting this data and making sure that your organization is aligned in terms of, ‘We’re going to have this approach to make sure that no participant data is being published’ and using those same anonymization approaches if you have, say, a submission in Japan or something like that,” he says. “But then from the earlier on approach too, making sure that if you are applying CCI redactions to documents that go into CTIS, a lot of those need to be translated, even the Part One documents, depending on which country you go into, you may need to supply that in a local language as well as English.
“So, we really work with sponsors trying to early on identify what is the CCI that [they] need, and we can apply the redactions to those English versions. But then you need to also be prepared to have certain documents, especially your Part Two country-specific documents, if there are any terms that you’re trying to keep out of the public eye to make sure that you’re aligned on that and have someone who’s able to read and translate and redact those terms in the local language versions as well.”
Zimmerman says the Real Life Sciences system allows for reuse of projects from one region to another. “So, a PRCI project, a Policy 0070 project, or CTR redactions can all be housed and managed within one system,” he says. “And then essentially you’re avoiding the need to rework and duplicate some of your work effort in terms of preparing these submission projects or these individual documents for redaction, because you can reuse them across studies, across projects if you will, for example, within from one CTR project to the next as it makes sense to do so. And then reuse across Policy 0070 and PRCI.
“Similarly for your voluntary data-sharing initiatives, if you’ve anonymized data for support of anonymizing your documents for Policy 0070 submission, you can then use that same anonymized dataset for other voluntary sharing purposes, whether that’s through platforms like Vivli or under a direct contract, a data-sharing agreement with a partner or an academic organization that you want to share the datasets with. So, there’s a lot of reuse opportunity here that helps you fulfill your overall objectives from a transparency perspective but minimizes your costs and your work effort.”
Shear says in theory a sponsor could switch to a deferral of the Policy 0070 submission if the EU member states do not agree with what has been redacted. “But I’m not sure it would work if you got the RFI from a member state and they said [the document] was over-redacted,” he says. “Or alternatively they said, ‘We don’t like the deferral period you selected,’ although we haven’t heard anything about them acting on deferrals yet.
“But in theory, you could go ahead and alter the application. You would have to change the structured data in terms of changing in the form section, what you would be deferring and for how long. You’d have to submit a summary of changes … in a free text field in terms of all the things that you changed and then you’d resubmit that and see if the member states were then satisfied. That in theory could work. I haven’t heard of anyone trying that yet, so I’m not sure if it absolutely would.”
Shear says since Policy 0070 was finalized in July, he has seen two different clients request a deferral, and they had redactions. “In one case, the redactions were so extensive that it was clearly going to get caught for over-redaction,” he says. “And they replied with an RFI of please cite the guidance, these documents are over-redacted and then rushed into a 12-day turnaround period of trying to redo all that work.”
In a theoretical scenario where a sponsor simultaneously redacts CCI while requesting a deferral for the same document, Shear says he had not seen that go through many times before the guidance was finalized. “So, member states might be becoming more strict on that, but the guidance is very clear that you should not be redacting and deferring, you should be doing one of those two approaches,” he says.
Zimmerman says for the first year and a half that CTIS was live, there wasn’t a lot of pushback in RFIs around over-redaction. “But it’s starting now, and multiple sponsors have cited this,” he says.
“You really need to be cautious because otherwise … I think everybody can appreciate the implications of a 12-day RFI turnaround are tough for any organization,” he adds. “And when you have to change strategy around how you’ve redacted these documents, you don’t have a lot of time to work with. So it’s really important to get it right up front and avoid those RFIs.”
Shear says he has seen one approach where companies create two versions of a redacted document: one they wish will be acceptable and one that’s more likely to be acceptable. Then, they have a second, less-aggressive document ready to submit if the first gets rejected.
“Sometimes they’re preparing these lists of redacted terms and you’ll say, ‘Here’s everything we wish we could redact,’” he says. “‘But if we get pushback, here’s just the few terms that we really need to.’
“In my opinion, a lot of this stuff is overdone. If you’re requesting a long deferral period, probably a lot of this information doesn’t need to be redacted anyway. So, you could be saving yourself a lot of work if you’re being a lot more honest with when are we going to publish this information, is this really unique to our product, and things like that.”





